
 PROTEIN PURIFICATION
PROTEIN PURIFICATION
Protein Purification by Ion-Exchange Chromatography
Ion exchange protein purification is possible because most proteins bear nonzero net electrostatic charges at all pHs except at pH=pI (isoelectric point). At a pH >pI of a given protein, that protein becomes negatively charged (an anion), at the pH<pI of that same protein, it becomes positively charged (a cation).
Ion exchange chromatography occurs due to electrostatic attraction between buffer-dissolved charged proteins and oppositely charged binding sites on a solid ion exchange adsorbent. An ion exchange adsorbent (also called media, resin, gel, or matrix) usually consists of spherical porous inert beads with charged groups (functional groups) densely grafted onto the beads' surfaces; the charges of functional groups are neutralized by free counter-ions.
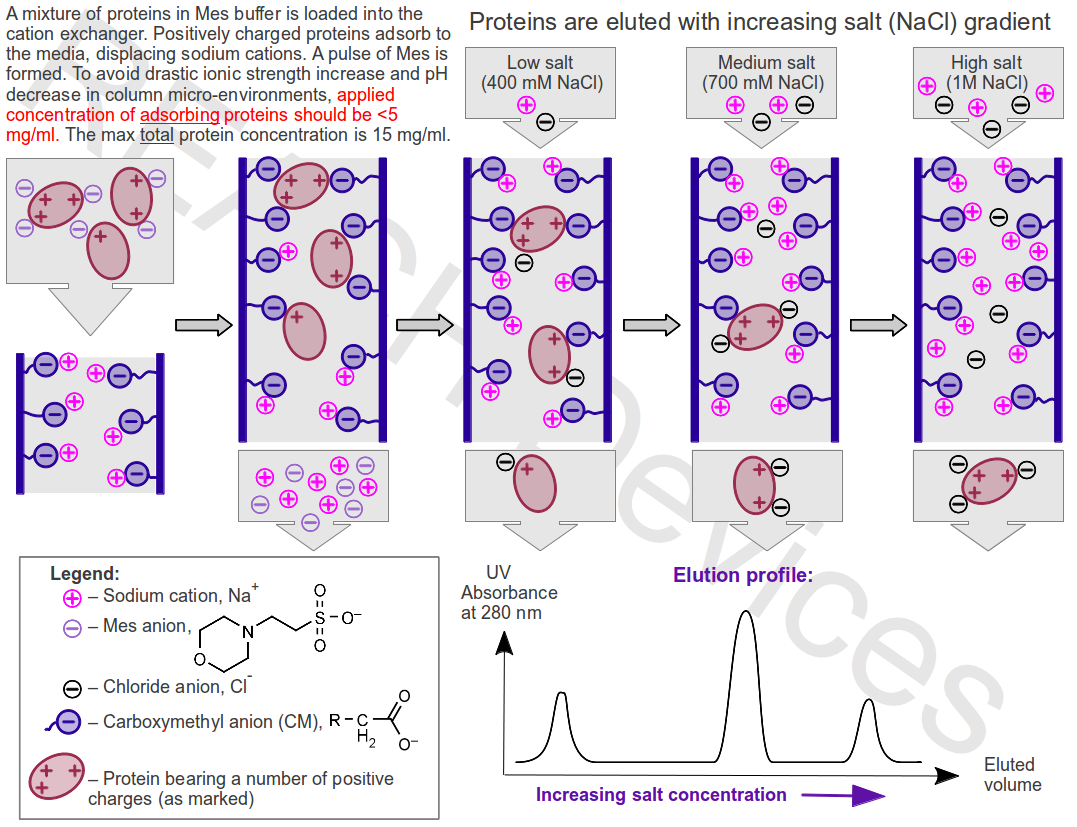
General steps for ion exchange chromatographic purification
- Protein mixture is transferred into low ionic strength buffer (mobile phase).
- Ion exchange adsorbent (stationary phase) is packed into a column, and the column is pre-equilibrated with the buffer of identical pH and similar ionic strength as protein mixture (preferably the same buffer as protein mixture).
- Protein mixture is applied onto the column. Proteins charged oppositely to ion-exchange media are temporarily retained in the column. All other proteins simply pass through the column and are collected during this step.
- Retained proteins are eluted from the column by applying a modified buffer. Elution is most commonly achieved by gradually increasing ionic strength of the buffer via salt gradient, and proteins are eluted in order of increasing their net charges. Is specific cases the elution can be accomplished by (a) pH change and (b) affinity methods.
Ion exchange chromatography can provide high-resolution separation for proteins with the same sign but various total net charge. Due to the high capacity of most ion-exchangers, the technique can also be used for capture of a mixture of same-sign charged proteins from large-volume diluted samples, the proteins are then eluted in considerably decreased sample volume.
What is happening during an ion exchange chromatographic purification
Application. During the application of the protein mixture onto the stationary phase, proteins adsorb from the solution to oppositely charged ion-exchanger media by displacing buffer ions that originally balanced charges on the functional groups. The adsorption is reversible and occurs in continuous competition for the binding sites between same-sign charged proteins and the buffer ions. In low ionic strength buffers, the concentration of competing buffer ions is low, and proteins spend most of their time adsorbed to binding sites on the stationary phase. However, even strongly bound proteins still spend some time in the solution, and therefore they continue to slowly move down the column with buffer flow. For example, if a given medium-bound protein spends 95% of the time adsorbed to the stationary phase, this protein will move down the column at 5% speed of the buffer and will emerge from the bottom of the column after 20 column volumes of buffer have been applied. Proteins bearing smaller electric charges move down the column faster then proteins bearing larger electric charges.
Elution with salt gradient. Addition of salt increases the number of ions competing with proteins for functional groups on the stationary phase. Proteins spend more time in the solution, the rate of their movement down the column increases dramatically, and proteins begin to elute from the column, usually in order of increasing charge. Most proteins are eluted at NaCl concentrations < 1M.
Elution by pH change. Change of pH in the column can be aimed to decrease the net absolute value of the charges of adsorbed proteins, decrease their attraction to the stationary phase, and accelerate the elution. In practice, pH changes in the column are difficult to control, as they do not reliably correspond to pH changes of the applied eluting buffer. This happens because of the buffering power of proteins adsorbed to the column and, for weak ion exchangers (see below), buffering power of the adsorbent functional groups themselves. Resolution of proteins by pH elution is achieved in a separate technique called “Chromatofocusing.”
Elution by affinity. Affinity elution can be achieved for a specific protein if and only if an oppositely charged ligand that will strongly bind to this protein is known and available. Addition of such a ligand to the eluting buffer will produce a protein+ligand species with a smaller absolute value of the net charge, and therefore the targeted protein will bind less to the stationary phase. Affinity elution is often useful in enzyme purifications.
Things to keep in mind
Before loading the protein mixture into the column, charged sites on proteins and ion exchanger functional groups are balanced by buffer counter-ions. When proteins adsorb to the ion exchangers, these buffer ions are released (a pulse of extra-buffer salts is formed): 1mg/ml of absorbed protein in general releases ~1mM of extra buffer salt. Therefore, while a part of the protein mixture is adsorbed, the remaining proteins in solution are exposed to an ionic strength increase (inhibits further adsorption), and pH changes (might cause protein damages).
| Adsorbent functional groups charge | Protein charge | Typical counter-ion balancing protein's charge |
Typical counter-ion balancing adsorbent charge |
What is released | Resulting pH change during loading |
| Positive (Anion Exchanger) | Negative | HTris+ | Cl- | HTris+ and Cl- (HTris+ = Tris + H+) | Decrease – more acidic |
| Negative (Cation Exchanger) | Positive | Mes- | Na+ | Mes- and Na+(Mes- + HOH = MesH + OH-) | Increase – more basic |
To minimize the impact of ionic strength and pH changes during loading:
- Applied concentration of adsorbing proteins should be < 5 mg/ml, while the total concentration of protein in the loaded mixture should not exceed 15 mg/ml.
- At least 10 mM buffers, ideally within 0.3 units of their pKa (at most within 0.5 unit of pKa), should be used during loading. The maximum buffer concentration should not exceed 50 mM.
Donnan effect.At any time, the actual pH in the micro-environments of ion-exchangers can be up to 1 pH unit different from the pH of the applied buffer. Care should be taken that proteins of interest are stable at the actual “resulting” pH.
- Negative functional groups of ion exchangers attract protons and repel hydroxyl anions from aqueous solutions, causing a more acidic pH in close proximity to protein binding sites.
- Positive functional groups of ion exchangers attract hydroxyl anions and repel protons from aqueous solutions, causing a more acidic pH in close proximity to protein binding sites.
Complications. In an ideal case, proteins adsorb to the solid ion-exchanger exclusively by electrostatic forces at the expected pH. In non-ideal, real world cases:
- Polyanionic complexing agents such as EDTA will bind to positively charged functional groups on anion exchangers, will accumulate on the solid phase, and so will preclude protein binding.
- Protein movement down the column can be slowed by hydrophobic attraction and hydrogen bonding of protein to the ion-exchangers' material. Hydrophobic attraction may cause irreversible binding or denaturation during the elution step. To lessen hydrophobic attractions, 10% of acetonitrile can be added to the buffer.
- Various surface charges on proteins can be unevenly distributed or inaccessible. In such cases, proteins may adsorb to cation exchangers at pH up to pI+1 (pH down to pI-1 for anion exchangers), or proteins, may not bind to the solid phase at the “expected” pH.
Nomenclature
In Cation-Exchange Chromatography, positively charged proteins (cations) are attracted to the negatively charged solid media (buffer cations originally balancing negative charges on the solid media are “exchanged” by protein cations).
In Anion-Exchange Chromatography, negatively charged proteins (anions) are attracted to the positively charged solid media (buffer anions originally balancing positive charges on the solid media are “exchanged” by protein anions).
| Ion exchange adsorbent type: | Cation Exchanger | Anion Exchanger |
| Functional groups of ion exchanger are charged: | Negatively | Positively |
| Protein's of interest net charge: | Positive (protein is a cation) | Negative (protein is an anion) |
| Advised buffer pH: | < pI - 1 | > pI + 1 |
| Resulting pH in the vicinity of solid phase (Donnan effect): | Up to 1 unit lower than buffer pH | Up to 1 unit higher than buffer pH |
Ion exchangers are usually additionally classified as “weak” or “strong.” The classification refers to the fact that functional groups on many ion-exchange absorbents maintain their charge only during a certain interval of pH. Functional groups on strong ion exchangers remain charged in wider pH windows than for weak ion exchangers. The weak/strong classification does not refer to the absorbent ability to bind proteins, it refers only to pKa value of their functional groups.
Buffer choice for ion exchange chromatography
Buffer choice can determine the success of ion exchange purification. The general nature of the buffer, concentration of the buffer, and the pKa of the buffer should all be considered. How all these properties relate to ion exchange chromatography is best understood after gaining a solid understanding of the concept of ionic strength of the buffer solution. Therefore, we will begin with a cursory overview of ionic strength.
Concept of ionic strength
An aqueous solution of any salt(s), including buffer salts, will have a certain value of ionic strength I. For a solution containing n types of ions numbered 1, 2,...n, charged z1, z2 … zn, at molar concentrations C1, C2,...Cn, the ionic strength I is defined as:
I = ½(C1*z12 + C2*z22 + … Cn*zn2) = ½∑Cizi2
Let's look at an example of 0.4M MgCl2 in water. Magnesium chloride fully dissociates according to equation:
MgCl2 = Mg+2 + 2Cl-
Thus, a 0.4M aqueous solution of MgCl2 will contain 0.4M Mg+2 ions and 0.8M of Cl- ions. The ionic strength of this solution is calculated as:
I = ½(CMg*zMg2 + CCl*zCl2) = ½(0.4*22 + 0.8*(-1)2) = 1.2
As it can be deduced from the above equations, as concentration of a salt increases, the ionic strength of the solution of this salt also increases. However, the exact relationship between the rates of increase of ionic strength and the concentration depends on the nature of the salt.
Nature and concentration of the buffer
Protein charge (both local and net), stability, and sometimes conformation are pH-dependent. As such, to avoid pH changes in column micro-environments, the charged form of the buffer should ideally have the same sign as the functional groups on the ion-exchanger.
Protein binding strength during ion exchange chromatography is pH-dependent, and decreases with increasing ionic strength of the buffer solution. A buffer provides a larger buffering capacity (stabilizing pH, and so protein binding strength) at a higher concentration of the buffer itself. An ideal buffer is one that provides a high buffering capacity while still not decreasing protein binding too much. As such, the best buffer is one whose ionic strength increases the slowest as buffer concentration increases. To minimize the ionic strength of the buffer:
- the buffer concentration should be 10-20mM,
- only one of the two buffering species should be charged, the other being neutral,
- the buffer should be prepared in such a way that water, not salt is formed.
These guidelines are summarized by the table below:
| Buffer type | In which ion exchange purification type the butter should ideally be used | Relationship between buffer concentration C and ionic strength I at pH=pKa | Preparation examples | On dilution, the buffer's pH and pKa are |
| HA = H+ + A-1 One neutral species |
Cation exchange – these are the best buffers | I ≈ 0.5*C With increasing buffer concentration C, the ionic strength I doesn't increase too much, which is good |
YES: CH3COOH + 0.5NaOH = 0.5CH3COOH + 0.5CH3COONa + 0.5H2O (water is formed) NO: CH3COONa + 0.5HCl = 0.5CH3COONa + 0.5CH3COOH + 0.5NaCl (salt is formed) |
Increased |
| HA-1 = H+ + A-2 Both species are charged |
Cation exchange – these are acceptable buffers | I ≈ 2*C With increasing buffer concentration C, the ionic strength I increases quite a bit, which is not optimal |
YES: NaH2PO4 + 0.5NaOH = 0.5NaH2PO4 + 0.5Na2HPO4 + 0.5H2O (water is formed) NO: Na2HPO4 + 0.5HCl = 0.5Na2HPO4 + 0.5NaH2PO4 + 0.5NaCl (salt is formed) |
Strongly increased |
| HA-2 = H+ + A-3 Both species are highly charged |
Cation exchange – these are barely acceptable buffers | I ≈ 4.5*C With increasing buffer concentration C, the ionic strength I increases dramatically, which is barely acceptable |
Very strongly increased | |
| HA+ = H+ + A One neutral species |
Anion exchange – these are the best buffers | I ≈ 0.5*C With increasing buffer concentration C, the ionic strength I doesn't increase too much, which is good |
YES: Tris + 0.5HCl = 0.5Tris + 0.5Tris*HCl + 0.5H2O (water is formed) NO: Tris*HCl + 0.5NaOH = 0.5Tris*HCl + 0.5Tris + 0.5NaCl (salt is formed) |
Decreased |
| HA+2 = H+ + A+ Both species are charged |
Anion exchange – these are acceptable buffers | I ≈ 2*C With increasing buffer concentration C, the ionic strength I increases quite a bit, which is not optimal |
Strongly decreased |
Please note that this information is a set of guidelines, and exceptions are possible. For example, phosphate buffer, H2PO4-1 = H+ + HPO4-2 (a type of HA-1 = H+ + A-2 buffer) has been used for anion exchange purification of enzymes due to those enzymes being stabilized best by said buffer.
pKa vs pKa0 of the buffer
The pKa of a buffer is commonly perceived as the pH of the said buffer when the concentrations of the two buffering species are equal, and where the maximum buffering capacity is achieved. However, it is often forgotten, that when defined as above, pKa depends on buffer concentration and temperature. To avoid this problem the concept of “thermodynamic” pKa0 was introduced. pKa0 is pKa of the buffer at infinite dilution (buffer concentration=0) and 25oC. Thus, pKa0 is a true constant specific for a given buffer.
In practice, this means that if one prepares the buffer concentrate by mixing of equal amounts of two buffering species, the buffer pH will indeed be equal to the buffer pKa (but not pKa0). Now, if one dilutes the said buffer concentrate, the solution will change its pH because pKa is changing with buffer concentration. If one further dilutes this buffer (infinite dilution at 25oC) buffer pH becomes equal pKa0.
Please note that, depending on the nature of the buffer, the pH of the buffer solution may increase or decrease upon dilution, and this effect may be significant. Additionally, small changes in temperature can also cause noticeable changes in the pH of the buffer solution.
Unfortunately, many tables of buffer pKas present mixed values of pKa and pKa0 without specifying any details.
The links below will transfer you to tables of buffers suitable for cation and anion exchange chromatography. For your convenience, these tables contain values of pKa0, d(pKa0)/dt at 298.25 K, and a calculator that allows you to estimate pKa values of each buffer at temperatures form 3oC (cold room) to 37oC, and concentrations from 1mM up to 500mM.
Recommended Buffers for Anion Exchange Chromatography
Recommended Buffers for Cation Exchange Chromatography
For a detailed explanation of pKa vs pKa0, and formulas used in the calculator, click on this link.
Chromatographic Materials for IE Protein Purifications
Traditionally, natural carbohydrate polymers – dextran, cellulose, agarose – have been widely used for protein purification. Their major advantage is their hydrophilic character that results in low, non-specific adsorption. Such polymers have low solid densities (>90% water) and limited mechanical strength, and therefore chemical cross-linking is used to achieve reasonable mechanical stability.
High mechanical stabilities can be achieved for synthetic polymers (higher solid densities, 50-80% water), however, all of them share a common disadvantage in protein purifications - relative hydrophobicity, increasing in order:
natural carbohydrate polymers < acrylamide/vinyl copolymers < methacrylate polymers < styrene/divinylbenzene copolymers.
Styrene/divinylbenzene-based beads are not suitable for many protein purifications due to high non-specific binding and low recovery. While attempts had been made to cover styrenic beads with hydrophilic coatings, we did not include styrene/divinylbenzene-based materials (Source, POROS, etc.) in the lists of commercially available ion-exchangers (links at the end of this page), as we feel such materials are better suited for applications other than protein purifications. We also avoided silica-based beads because of their low stability under alkaline conditions.
| Ion Exchanger type |
Functional group on IE matrix | Charge Z of the functional groups | ||||||||||
| Abbreviation | Name | Structure | pKa | |||||||||
| pH of the solution | ||||||||||||
| <2 | 2-2.5 | 2.5-3 | 3-5 | 5-7 | 7-10 | 10-11 | >11 | |||||
| Strong anion exchanger | Q | Trimethyl aminomethyl | -O-CH2-N+(CH3)3 | Z=+1 | Not stable | |||||||
| TMAE | Trimethyl aminoethyl | -O-CH2-CH2-N+(CH3)3 | ||||||||||
| QAE | Diethyl-(2-hydroxypropyl) aminoethyl | -O-CH2-CH2-N+(CH2-CH3)2(CH2-CHOH-CH3) | ||||||||||
| QA | Trimethylamino-hydroxypropyl | -O-CH2-CHOH-CH2-N+(CH3)3 | ||||||||||
| Weak anion exchanger | DMAE | Dimethyl aminoethyl | -O-CH2-CH2-NH+(CH3)2 | ~10 | Z=+1 | 0<Z<1 | Z=0 | |||||
| Weak anion exchanger | DEAE | Diethyl aminoethyl | -O-CH2-CH2-NH+(CH2-CH3)2 | 6-9* | Z=+1 | 0<Z<1 | Z=0 | |||||
| Weak cation exchanger | CM | Carboxymethyl | -O-CH2-COO- | 3.5-4.5 | Z~0 | -1<Z<0 | Z=-1 | |||||
| Strong cation exchanger | SP | Sulfopropyl | -O-CH2-CH2-CH2-SO3- | 2-2.5 | Z~0 | Z=-1 | ||||||
| SE | Sulfoethyl | -O-CH2-CH2-SO3- | 2 | Z~0 | Z=-1 | |||||||
| S | Sulfomethyl | -O-CH2-SO3- | 2 | |||||||||
REACH Devices, LLC.
6525 Gunpark Drive, Suite 370-179, Boulder, CO 80301
Call: (720) 288-5722
Inquiries: support@reachdevices.com
Ⓒ 2010-2025 REACH Devices, LLC. All rights reserved.